
Samuel Sledzieski
commited on
Commit
•
e09f17f
1
Parent(s):
d43f920
add tt3d with prostt5 predictino of 3di sequences
Browse files- .gitignore +6 -0
- app.py +123 -21
- dscript_architecture1.png +0 -0
- predict_3di.py +354 -0
- requirements.txt +2 -0
.gitignore
ADDED
|
@@ -0,0 +1,6 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
__pycache__/
|
| 2 |
+
models
|
| 3 |
+
cnn_chkpnt
|
| 4 |
+
foldseek
|
| 5 |
+
*.fasta
|
| 6 |
+
*.tar.gz
|
app.py
CHANGED
|
@@ -1,52 +1,151 @@
|
|
|
|
|
|
|
|
| 1 |
import gradio as gr
|
| 2 |
import pandas as pd
|
|
|
|
| 3 |
from pathlib import Path
|
| 4 |
from Bio import SeqIO
|
| 5 |
from dscript.pretrained import get_pretrained
|
| 6 |
from dscript.language_model import lm_embed
|
| 7 |
from tqdm.auto import tqdm
|
| 8 |
from uuid import uuid4
|
|
|
|
| 9 |
|
| 10 |
model_map = {
|
| 11 |
"D-SCRIPT": "human_v1",
|
| 12 |
-
"Topsy-Turvy": "human_v2"
|
|
|
|
| 13 |
}
|
| 14 |
|
| 15 |
-
|
| 16 |
-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 17 |
run_id = uuid4()
|
|
|
|
| 18 |
|
| 19 |
-
gr.Info("Loading model...")
|
| 20 |
-
_ = lm_embed("M")
|
| 21 |
|
| 22 |
-
model = get_pretrained(model_map[
|
| 23 |
|
| 24 |
-
gr.Info("Loading files...")
|
| 25 |
try:
|
| 26 |
seqs = SeqIO.to_dict(SeqIO.parse(sequence_file.name, "fasta"))
|
| 27 |
-
except ValueError as
|
| 28 |
-
gr.Error("Invalid FASTA file - duplicate entry")
|
| 29 |
|
| 30 |
if Path(pairs_file.name).suffix == ".csv":
|
| 31 |
pairs = pd.read_csv(pairs_file.name)
|
| 32 |
elif Path(pairs_file.name).suffix == ".tsv":
|
| 33 |
pairs = pd.read_csv(pairs_file.name, sep="\t")
|
| 34 |
-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 35 |
|
| 36 |
-
|
| 37 |
results = []
|
| 38 |
-
|
| 39 |
-
|
| 40 |
-
|
|
|
|
| 41 |
prot1 = r["protein1"]
|
| 42 |
prot2 = r["protein2"]
|
|
|
|
| 43 |
seq1 = str(seqs[prot1].seq)
|
| 44 |
seq2 = str(seqs[prot2].seq)
|
|
|
|
|
|
|
|
|
|
|
|
|
| 45 |
lm1 = lm_embed(seq1)
|
| 46 |
lm2 = lm_embed(seq2)
|
| 47 |
-
|
|
|
|
|
|
|
|
|
|
| 48 |
results.append([prot1, prot2, interaction])
|
| 49 |
-
progress((i, len(pairs)))
|
| 50 |
|
| 51 |
results = pd.DataFrame(results, columns = ["Protein 1", "Protein 2", "Interaction"])
|
| 52 |
|
|
@@ -59,16 +158,19 @@ def predict(model, sequence_file, pairs_file):
|
|
| 59 |
demo = gr.Interface(
|
| 60 |
fn=predict,
|
| 61 |
inputs = [
|
| 62 |
-
gr.Dropdown(label="Model", choices = ["D-SCRIPT", "Topsy-Turvy"], value = "Topsy-Turvy"),
|
|
|
|
| 63 |
gr.File(label="Sequences (.fasta)", file_types = [".fasta"]),
|
| 64 |
-
gr.File(label="Pairs (.csv/.tsv)", file_types = [".csv", ".tsv"])
|
| 65 |
],
|
| 66 |
outputs = [
|
| 67 |
gr.DataFrame(label='Results', headers=['Protein 1', 'Protein 2', 'Interaction']),
|
| 68 |
gr.File(label="Download results", type="file")
|
| 69 |
-
]
|
|
|
|
|
|
|
|
|
|
|
|
|
| 70 |
)
|
| 71 |
|
| 72 |
if __name__ == "__main__":
|
| 73 |
-
demo.queue(max_size=20)
|
| 74 |
-
demo.launch()
|
|
|
|
| 1 |
+
import time
|
| 2 |
+
|
| 3 |
import gradio as gr
|
| 4 |
import pandas as pd
|
| 5 |
+
import torch
|
| 6 |
from pathlib import Path
|
| 7 |
from Bio import SeqIO
|
| 8 |
from dscript.pretrained import get_pretrained
|
| 9 |
from dscript.language_model import lm_embed
|
| 10 |
from tqdm.auto import tqdm
|
| 11 |
from uuid import uuid4
|
| 12 |
+
from predict_3di import get_3di_sequences, predictions_to_dict, one_hot_3di_sequence
|
| 13 |
|
| 14 |
model_map = {
|
| 15 |
"D-SCRIPT": "human_v1",
|
| 16 |
+
"Topsy-Turvy": "human_v2",
|
| 17 |
+
"TT3D": "human_tt3d",
|
| 18 |
}
|
| 19 |
|
| 20 |
+
theme = "Default"
|
| 21 |
+
title = "D-SCRIPT: Predicting Protein-Protein Interactions"
|
| 22 |
+
description = """
|
| 23 |
+
"""
|
| 24 |
+
|
| 25 |
+
article = """
|
| 26 |
+
|
| 27 |
+
<hr>
|
| 28 |
+
|
| 29 |
+
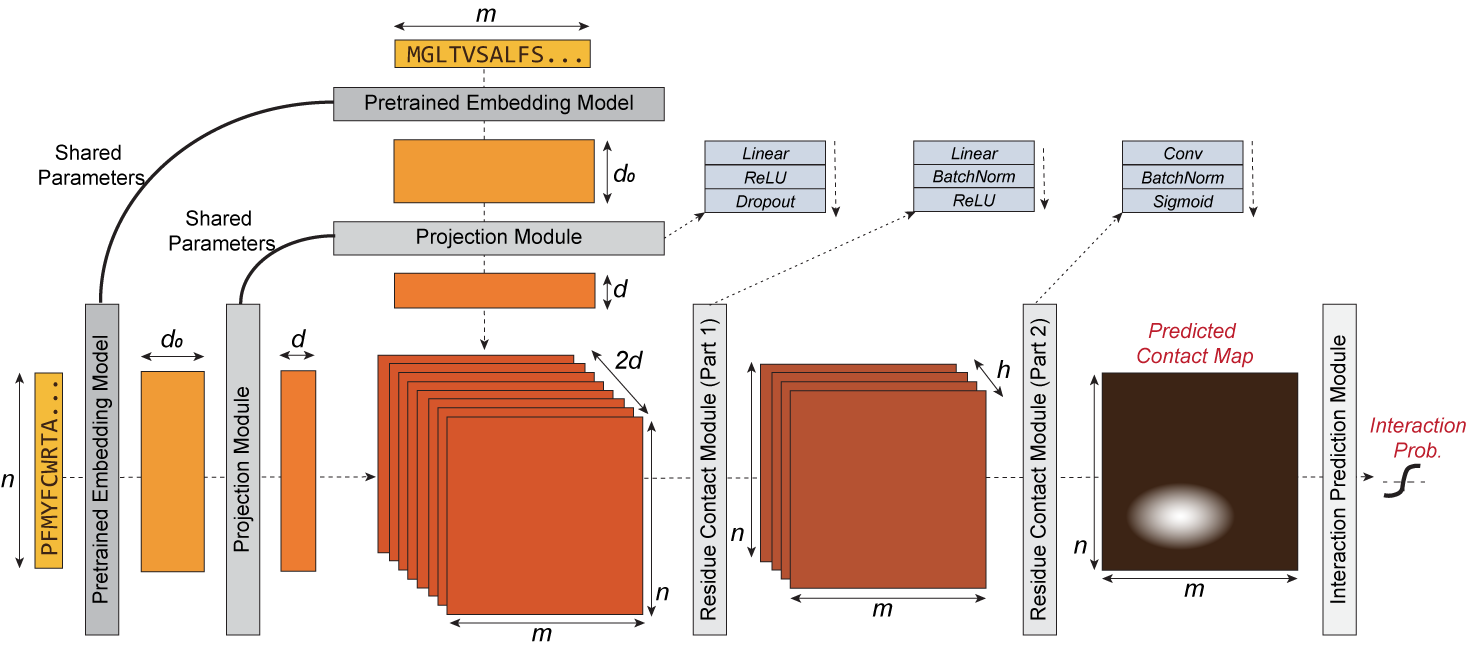
<img style="margin-left:auto; margin-right:auto" src="https://raw.githubusercontent.com/samsledje/D-SCRIPT/main/docs/source/img/dscript_architecture.png" alt="D-SCRIPT architecture" width="70%"/>
|
| 30 |
+
|
| 31 |
+
<hr>
|
| 32 |
+
|
| 33 |
+
D-SCRIPT is a deep learning method for predicting a physical interaction between two proteins given just their sequences.
|
| 34 |
+
It generalizes well to new species and is robust to limitations in training data size. Its design reflects the intuition that for two proteins to physically interact,
|
| 35 |
+
a subset of amino acids from each protein should be in contact with the other. The intermediate stages of D-SCRIPT directly implement this intuition, with the penultimate stage
|
| 36 |
+
in D-SCRIPT being a rough estimate of the inter-protein contact map of the protein dimer. This structurally-motivated design enhances the interpretability of the results and,
|
| 37 |
+
since structure is more conserved evolutionarily than sequence, improves generalizability across species.
|
| 38 |
+
|
| 39 |
+
<hr>
|
| 40 |
+
|
| 41 |
+
Computational methods to predict protein-protein interaction (PPI) typically segregate into sequence-based "bottom-up" methods that infer properties from the characteristics of the
|
| 42 |
+
individual protein sequences, or global "top-down" methods that infer properties from the pattern of already known PPIs in the species of interest. However, a way to incorporate
|
| 43 |
+
top-down insights into sequence-based bottom-up PPI prediction methods has been elusive. Topsy-Turvy builds upon D-SCRIPT by synthesizing both views in a sequence-based,
|
| 44 |
+
multi-scale, deep-learning model for PPI prediction. While Topsy-Turvy makes predictions using only sequence data, during the training phase it takes a transfer-learning approach by
|
| 45 |
+
incorporating patterns from both global and molecular-level views of protein interaction. In a cross-species context, we show it achieves state-of-the-art performance, offering the
|
| 46 |
+
ability to perform genome-scale, interpretable PPI prediction for non-model organisms with no existing experimental PPI data.
|
| 47 |
+
|
| 48 |
+
|
| 49 |
+
"""
|
| 50 |
+
|
| 51 |
+
fold_vocab = {
|
| 52 |
+
"D": 0,
|
| 53 |
+
"P": 1,
|
| 54 |
+
"V": 2,
|
| 55 |
+
"Q": 3,
|
| 56 |
+
"A": 4,
|
| 57 |
+
"W": 5,
|
| 58 |
+
"K": 6,
|
| 59 |
+
"E": 7,
|
| 60 |
+
"I": 8,
|
| 61 |
+
"T": 9,
|
| 62 |
+
"L": 10,
|
| 63 |
+
"F": 11,
|
| 64 |
+
"G": 12,
|
| 65 |
+
"S": 13,
|
| 66 |
+
"M": 14,
|
| 67 |
+
"H": 15,
|
| 68 |
+
"C": 16,
|
| 69 |
+
"R": 17,
|
| 70 |
+
"Y": 18,
|
| 71 |
+
"N": 19,
|
| 72 |
+
"X": 20,
|
| 73 |
+
}
|
| 74 |
+
|
| 75 |
+
def predict(model_name, pairs_file, sequence_file, progress = gr.Progress()):
|
| 76 |
+
|
| 77 |
run_id = uuid4()
|
| 78 |
+
device = torch.cuda("0") if torch.cuda.is_available() else torch.device("cpu")
|
| 79 |
|
| 80 |
+
# gr.Info("Loading model...")
|
| 81 |
+
_ = lm_embed("M", use_cuda = (device.type == "cuda"))
|
| 82 |
|
| 83 |
+
model = get_pretrained(model_map[model_name]).to(device)
|
| 84 |
|
| 85 |
+
# gr.Info("Loading files...")
|
| 86 |
try:
|
| 87 |
seqs = SeqIO.to_dict(SeqIO.parse(sequence_file.name, "fasta"))
|
| 88 |
+
except ValueError as _:
|
| 89 |
+
raise gr.Error("Invalid FASTA file - duplicate entry")
|
| 90 |
|
| 91 |
if Path(pairs_file.name).suffix == ".csv":
|
| 92 |
pairs = pd.read_csv(pairs_file.name)
|
| 93 |
elif Path(pairs_file.name).suffix == ".tsv":
|
| 94 |
pairs = pd.read_csv(pairs_file.name, sep="\t")
|
| 95 |
+
try:
|
| 96 |
+
pairs.columns = ["protein1", "protein2"]
|
| 97 |
+
except ValueError as _:
|
| 98 |
+
raise gr.Error("Invalid pairs file - must have two columns 'protein1' and 'protein2'")
|
| 99 |
+
|
| 100 |
+
do_foldseek = False
|
| 101 |
+
if model_name == "TT3D":
|
| 102 |
+
do_foldseek = True
|
| 103 |
+
|
| 104 |
+
need_to_translate = set(pairs["protein1"]).union(set(pairs["protein2"]))
|
| 105 |
+
seqs_to_translate = {k: str(seqs[k].seq) for k in need_to_translate if k in seqs}
|
| 106 |
+
|
| 107 |
+
half_precision = False
|
| 108 |
+
assert not (half_precision and device=="cpu"), print("Running fp16 on CPU is not supported, yet")
|
| 109 |
+
|
| 110 |
+
gr.Info(f"Loading Foldseek embeddings -- this may take some time ({len(seqs_to_translate)} embeddings)...")
|
| 111 |
+
predictions = get_3di_sequences(
|
| 112 |
+
seqs_to_translate,
|
| 113 |
+
model_dir = "Rostlab/ProstT5",
|
| 114 |
+
report_fn = gr.Info,
|
| 115 |
+
error_fn = gr.Error,
|
| 116 |
+
device=device,
|
| 117 |
+
)
|
| 118 |
+
foldseek_sequences = predictions_to_dict(predictions)
|
| 119 |
+
foldseek_embeddings = {k: one_hot_3di_sequence(s.upper(), fold_vocab) for k, s in foldseek_sequences.items()}
|
| 120 |
+
|
| 121 |
+
# for k in seqs_to_translate.keys():
|
| 122 |
+
# print(seqs_to_translate[k])
|
| 123 |
+
# print(len(seqs_to_translate[k]))
|
| 124 |
+
# print(foldseek_embeddings[k])
|
| 125 |
+
# print(foldseek_embeddings[k].shape)
|
| 126 |
|
| 127 |
+
progress(0, desc="Starting...")
|
| 128 |
results = []
|
| 129 |
+
for i in progress.tqdm(range(len(pairs))):
|
| 130 |
+
|
| 131 |
+
r = pairs.iloc[i]
|
| 132 |
+
|
| 133 |
prot1 = r["protein1"]
|
| 134 |
prot2 = r["protein2"]
|
| 135 |
+
|
| 136 |
seq1 = str(seqs[prot1].seq)
|
| 137 |
seq2 = str(seqs[prot2].seq)
|
| 138 |
+
|
| 139 |
+
fold1 = foldseek_embeddings[prot1] if do_foldseek else None
|
| 140 |
+
fold2 = foldseek_embeddings[prot2] if do_foldseek else None
|
| 141 |
+
|
| 142 |
lm1 = lm_embed(seq1)
|
| 143 |
lm2 = lm_embed(seq2)
|
| 144 |
+
|
| 145 |
+
print(lm1.shape, lm2.shape, fold1.shape, fold2.shape)
|
| 146 |
+
interaction = model.predict(lm1, lm2, embed_foldseek = do_foldseek, f0 = fold1, f1 = fold2).item()
|
| 147 |
+
|
| 148 |
results.append([prot1, prot2, interaction])
|
|
|
|
| 149 |
|
| 150 |
results = pd.DataFrame(results, columns = ["Protein 1", "Protein 2", "Interaction"])
|
| 151 |
|
|
|
|
| 158 |
demo = gr.Interface(
|
| 159 |
fn=predict,
|
| 160 |
inputs = [
|
| 161 |
+
gr.Dropdown(label="Model", choices = ["D-SCRIPT", "Topsy-Turvy", "TT3D"], value = "Topsy-Turvy"),
|
| 162 |
+
gr.File(label="Pairs (.csv/.tsv)", file_types = [".csv", ".tsv"]),
|
| 163 |
gr.File(label="Sequences (.fasta)", file_types = [".fasta"]),
|
|
|
|
| 164 |
],
|
| 165 |
outputs = [
|
| 166 |
gr.DataFrame(label='Results', headers=['Protein 1', 'Protein 2', 'Interaction']),
|
| 167 |
gr.File(label="Download results", type="file")
|
| 168 |
+
],
|
| 169 |
+
title = title,
|
| 170 |
+
description = description,
|
| 171 |
+
article = article,
|
| 172 |
+
theme = theme,
|
| 173 |
)
|
| 174 |
|
| 175 |
if __name__ == "__main__":
|
| 176 |
+
demo.queue(max_size=20).launch()
|
|
|
dscript_architecture1.png
ADDED
|
predict_3di.py
ADDED
|
@@ -0,0 +1,354 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python3
|
| 2 |
+
# -*- coding: utf-8 -*-
|
| 3 |
+
"""
|
| 4 |
+
Created on Fri Jun 16 14:27:44 2023
|
| 5 |
+
|
| 6 |
+
@author: mheinzinger
|
| 7 |
+
"""
|
| 8 |
+
|
| 9 |
+
import argparse
|
| 10 |
+
import time
|
| 11 |
+
from pathlib import Path
|
| 12 |
+
|
| 13 |
+
from urllib import request
|
| 14 |
+
import shutil
|
| 15 |
+
|
| 16 |
+
import numpy as np
|
| 17 |
+
import torch
|
| 18 |
+
from torch import nn
|
| 19 |
+
from transformers import T5EncoderModel, T5Tokenizer
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
device = torch.device('cuda:0' if torch.cuda.is_available() else 'cpu')
|
| 23 |
+
print("Using device: {}".format(device))
|
| 24 |
+
|
| 25 |
+
|
| 26 |
+
# Convolutional neural network (two convolutional layers)
|
| 27 |
+
class CNN(nn.Module):
|
| 28 |
+
def __init__( self ):
|
| 29 |
+
super(CNN, self).__init__()
|
| 30 |
+
|
| 31 |
+
self.classifier = nn.Sequential(
|
| 32 |
+
nn.Conv2d(1024, 32, kernel_size=(7, 1), padding=(3, 0)), # 7x32
|
| 33 |
+
nn.ReLU(),
|
| 34 |
+
nn.Dropout(0.0),
|
| 35 |
+
nn.Conv2d(32, 20, kernel_size=(7, 1), padding=(3, 0))
|
| 36 |
+
)
|
| 37 |
+
|
| 38 |
+
def forward(self, x):
|
| 39 |
+
"""
|
| 40 |
+
L = protein length
|
| 41 |
+
B = batch-size
|
| 42 |
+
F = number of features (1024 for embeddings)
|
| 43 |
+
N = number of classes (20 for 3Di)
|
| 44 |
+
"""
|
| 45 |
+
x = x.permute(0, 2, 1).unsqueeze(dim=-1) # IN: X = (B x L x F); OUT: (B x F x L, 1)
|
| 46 |
+
Yhat = self.classifier(x) # OUT: Yhat_consurf = (B x N x L x 1)
|
| 47 |
+
Yhat = Yhat.squeeze(dim=-1) # IN: (B x N x L x 1); OUT: ( B x L x N )
|
| 48 |
+
return Yhat
|
| 49 |
+
|
| 50 |
+
def one_hot_3di_sequence(sequence, vocab):
|
| 51 |
+
foldseek_enc = torch.zeros(
|
| 52 |
+
len(sequence), len(vocab), dtype=torch.float32
|
| 53 |
+
)
|
| 54 |
+
for i, a in enumerate(sequence):
|
| 55 |
+
assert a in vocab
|
| 56 |
+
foldseek_enc[i, vocab[a]] = 1
|
| 57 |
+
return foldseek_enc.unsqueeze(0)
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
def get_T5_model(model_dir):
|
| 61 |
+
print("Loading T5 from: {}".format(model_dir))
|
| 62 |
+
model = T5EncoderModel.from_pretrained(model_dir).to(device)
|
| 63 |
+
model = model.eval()
|
| 64 |
+
vocab = T5Tokenizer.from_pretrained(model_dir, do_lower_case=False )
|
| 65 |
+
return model, vocab
|
| 66 |
+
|
| 67 |
+
|
| 68 |
+
def read_fasta( fasta_path, split_char, id_field ):
|
| 69 |
+
'''
|
| 70 |
+
Reads in fasta file containing multiple sequences.
|
| 71 |
+
Returns dictionary of holding multiple sequences or only single
|
| 72 |
+
sequence, depending on input file.
|
| 73 |
+
'''
|
| 74 |
+
|
| 75 |
+
sequences = dict()
|
| 76 |
+
with open( fasta_path, 'r' ) as fasta_f:
|
| 77 |
+
for line in fasta_f:
|
| 78 |
+
# get uniprot ID from header and create new entry
|
| 79 |
+
if line.startswith('>'):
|
| 80 |
+
uniprot_id = line.replace('>', '').strip().split(split_char)[id_field]
|
| 81 |
+
# replace tokens that are mis-interpreted when loading h5
|
| 82 |
+
uniprot_id = uniprot_id.replace("/","_").replace(".","_")
|
| 83 |
+
sequences[ uniprot_id ] = ''
|
| 84 |
+
else:
|
| 85 |
+
s = ''.join( line.split() ).replace("-","")
|
| 86 |
+
|
| 87 |
+
if s.islower(): # sanity check to avoid mix-up of 3Di and AA input
|
| 88 |
+
print("The input file was in lower-case which indicates 3Di-input." +
|
| 89 |
+
"This predictor only operates on amino-acid-input (upper-case)." +
|
| 90 |
+
"Exiting now ..."
|
| 91 |
+
)
|
| 92 |
+
return None
|
| 93 |
+
else:
|
| 94 |
+
sequences[ uniprot_id ] += s
|
| 95 |
+
return sequences
|
| 96 |
+
|
| 97 |
+
def write_predictions(predictions, out_path):
|
| 98 |
+
ss_mapping = {
|
| 99 |
+
0: "A",
|
| 100 |
+
1: "C",
|
| 101 |
+
2: "D",
|
| 102 |
+
3: "E",
|
| 103 |
+
4: "F",
|
| 104 |
+
5: "G",
|
| 105 |
+
6: "H",
|
| 106 |
+
7: "I",
|
| 107 |
+
8: "K",
|
| 108 |
+
9: "L",
|
| 109 |
+
10: "M",
|
| 110 |
+
11: "N",
|
| 111 |
+
12: "P",
|
| 112 |
+
13: "Q",
|
| 113 |
+
14: "R",
|
| 114 |
+
15: "S",
|
| 115 |
+
16: "T",
|
| 116 |
+
17: "V",
|
| 117 |
+
18: "W",
|
| 118 |
+
19: "Y"
|
| 119 |
+
}
|
| 120 |
+
|
| 121 |
+
with open(out_path, 'w+') as out_f:
|
| 122 |
+
out_f.write( '\n'.join(
|
| 123 |
+
[ ">{}\n{}".format(
|
| 124 |
+
seq_id, "".join(list(map(lambda yhat: ss_mapping[int(yhat)], yhats))) )
|
| 125 |
+
for seq_id, yhats in predictions.items()
|
| 126 |
+
]
|
| 127 |
+
) )
|
| 128 |
+
print(f"Finished writing results to {out_path}")
|
| 129 |
+
return None
|
| 130 |
+
|
| 131 |
+
def predictions_to_dict(predictions):
|
| 132 |
+
ss_mapping = {
|
| 133 |
+
0: "A",
|
| 134 |
+
1: "C",
|
| 135 |
+
2: "D",
|
| 136 |
+
3: "E",
|
| 137 |
+
4: "F",
|
| 138 |
+
5: "G",
|
| 139 |
+
6: "H",
|
| 140 |
+
7: "I",
|
| 141 |
+
8: "K",
|
| 142 |
+
9: "L",
|
| 143 |
+
10: "M",
|
| 144 |
+
11: "N",
|
| 145 |
+
12: "P",
|
| 146 |
+
13: "Q",
|
| 147 |
+
14: "R",
|
| 148 |
+
15: "S",
|
| 149 |
+
16: "T",
|
| 150 |
+
17: "V",
|
| 151 |
+
18: "W",
|
| 152 |
+
19: "Y"
|
| 153 |
+
}
|
| 154 |
+
|
| 155 |
+
results = {seq_id: "".join(list(map(lambda yhat: ss_mapping[int(yhat)], yhats))) for seq_id, yhats in predictions.items()}
|
| 156 |
+
return results
|
| 157 |
+
|
| 158 |
+
def toCPU(tensor):
|
| 159 |
+
if len(tensor.shape) > 1:
|
| 160 |
+
return tensor.detach().cpu().squeeze(dim=-1).numpy()
|
| 161 |
+
else:
|
| 162 |
+
return tensor.detach().cpu().numpy()
|
| 163 |
+
|
| 164 |
+
|
| 165 |
+
def download_file(url,local_path):
|
| 166 |
+
if not local_path.parent.is_dir():
|
| 167 |
+
local_path.parent.mkdir()
|
| 168 |
+
|
| 169 |
+
print("Downloading: {}".format(url))
|
| 170 |
+
req = request.Request(url, headers={
|
| 171 |
+
'User-Agent' : 'Mozilla/5.0 (Windows NT 6.1; Win64; x64)'
|
| 172 |
+
})
|
| 173 |
+
|
| 174 |
+
with request.urlopen(req) as response, open(local_path, 'wb') as outfile:
|
| 175 |
+
shutil.copyfileobj(response, outfile)
|
| 176 |
+
return None
|
| 177 |
+
|
| 178 |
+
|
| 179 |
+
def load_predictor( weights_link="https://rostlab.org/~deepppi/prostt5/cnn_chkpnt/model.pt" , device=torch.device("cpu")):
|
| 180 |
+
model = CNN()
|
| 181 |
+
checkpoint_p = Path.cwd() / "cnn_chkpnt" / "model.pt"
|
| 182 |
+
# if no pre-trained model is available, yet --> download it
|
| 183 |
+
if not checkpoint_p.exists():
|
| 184 |
+
download_file(weights_link, checkpoint_p)
|
| 185 |
+
|
| 186 |
+
state = torch.load(checkpoint_p, map_location=device)
|
| 187 |
+
|
| 188 |
+
model.load_state_dict(state["state_dict"])
|
| 189 |
+
|
| 190 |
+
model = model.eval()
|
| 191 |
+
model = model.to(device)
|
| 192 |
+
|
| 193 |
+
return model
|
| 194 |
+
|
| 195 |
+
|
| 196 |
+
def get_3di_sequences( seq_dict, model_dir, device,
|
| 197 |
+
max_residues=4000, max_seq_len=1000, max_batch=100,report_fn=print,error_fn=print,half_precision=False):
|
| 198 |
+
|
| 199 |
+
predictions = dict()
|
| 200 |
+
|
| 201 |
+
prefix = "<AA2fold>"
|
| 202 |
+
|
| 203 |
+
model, vocab = get_T5_model(model_dir)
|
| 204 |
+
predictor = load_predictor(device=device)
|
| 205 |
+
|
| 206 |
+
if half_precision:
|
| 207 |
+
model = model.half()
|
| 208 |
+
predictor = predictor.half()
|
| 209 |
+
|
| 210 |
+
report_fn('Total number of sequences: {}'.format(len(seq_dict)))
|
| 211 |
+
|
| 212 |
+
avg_length = sum([ len(seq) for _, seq in seq_dict.items()]) / len(seq_dict)
|
| 213 |
+
n_long = sum([ 1 for _, seq in seq_dict.items() if len(seq)>max_seq_len])
|
| 214 |
+
# sort sequences by length to trigger OOM at the beginning
|
| 215 |
+
seq_dict = sorted( seq_dict.items(), key=lambda kv: len( seq_dict[kv[0]] ), reverse=True )
|
| 216 |
+
|
| 217 |
+
report_fn("Average sequence length: {}".format(avg_length))
|
| 218 |
+
report_fn("Number of sequences >{}: {}".format(max_seq_len, n_long))
|
| 219 |
+
|
| 220 |
+
start = time.time()
|
| 221 |
+
batch = list()
|
| 222 |
+
for seq_idx, (pdb_id, seq) in enumerate(seq_dict,1):
|
| 223 |
+
# replace non-standard AAs
|
| 224 |
+
seq = seq.replace('U','X').replace('Z','X').replace('O','X')
|
| 225 |
+
seq_len = len(seq)
|
| 226 |
+
seq = prefix + ' ' + ' '.join(list(seq))
|
| 227 |
+
batch.append((pdb_id,seq,seq_len))
|
| 228 |
+
|
| 229 |
+
# count residues in current batch and add the last sequence length to
|
| 230 |
+
# avoid that batches with (n_res_batch > max_residues) get processed
|
| 231 |
+
n_res_batch = sum([ s_len for _, _, s_len in batch ]) + seq_len
|
| 232 |
+
if len(batch) >= max_batch or n_res_batch>=max_residues or seq_idx==len(seq_dict) or seq_len>max_seq_len:
|
| 233 |
+
pdb_ids, seqs, seq_lens = zip(*batch)
|
| 234 |
+
batch = list()
|
| 235 |
+
|
| 236 |
+
token_encoding = vocab.batch_encode_plus(seqs,
|
| 237 |
+
add_special_tokens=True,
|
| 238 |
+
padding="longest",
|
| 239 |
+
return_tensors='pt'
|
| 240 |
+
).to(device)
|
| 241 |
+
try:
|
| 242 |
+
with torch.no_grad():
|
| 243 |
+
embedding_repr = model(token_encoding.input_ids,
|
| 244 |
+
attention_mask=token_encoding.attention_mask
|
| 245 |
+
)
|
| 246 |
+
except RuntimeError:
|
| 247 |
+
error_fn("RuntimeError during embedding for {} (L={})".format(
|
| 248 |
+
pdb_id, seq_len)
|
| 249 |
+
)
|
| 250 |
+
continue
|
| 251 |
+
|
| 252 |
+
# ProtT5 appends a special tokens at the end of each sequence
|
| 253 |
+
# Mask this also out during inference while taking into account the prefix
|
| 254 |
+
for idx, s_len in enumerate(seq_lens):
|
| 255 |
+
token_encoding.attention_mask[idx,s_len+1] = 0
|
| 256 |
+
|
| 257 |
+
# extract last hidden states (=embeddings)
|
| 258 |
+
residue_embedding = embedding_repr.last_hidden_state.detach()
|
| 259 |
+
# mask out padded elements in the attention output (can be non-zero) for further processing/prediction
|
| 260 |
+
residue_embedding = residue_embedding*token_encoding.attention_mask.unsqueeze(dim=-1)
|
| 261 |
+
# slice off embedding of special token prepended before to each sequence
|
| 262 |
+
residue_embedding = residue_embedding[:,1:]
|
| 263 |
+
|
| 264 |
+
prediction = predictor(residue_embedding)
|
| 265 |
+
prediction = toCPU(torch.max( prediction, dim=1, keepdim=True )[1] ).astype(np.byte)
|
| 266 |
+
|
| 267 |
+
# batch-size x seq_len x embedding_dim
|
| 268 |
+
# extra token is added at the end of the seq
|
| 269 |
+
for batch_idx, identifier in enumerate(pdb_ids):
|
| 270 |
+
s_len = seq_lens[batch_idx]
|
| 271 |
+
# slice off padding and special token appended to the end of the sequence
|
| 272 |
+
predictions[identifier] = prediction[batch_idx,:, 0:s_len].squeeze()
|
| 273 |
+
assert s_len == len(predictions[identifier]), error_fn(f"Length mismatch for {identifier}: is:{len(predictions[identifier])} vs should:{s_len}")
|
| 274 |
+
|
| 275 |
+
end = time.time()
|
| 276 |
+
report_fn('Total number of predictions: {}'.format(len(predictions)))
|
| 277 |
+
report_fn('Total time: {:.2f}[s]; time/prot: {:.4f}[s]; avg. len= {:.2f}'.format(
|
| 278 |
+
end-start, (end-start)/len(predictions), avg_length))
|
| 279 |
+
|
| 280 |
+
return predictions
|
| 281 |
+
|
| 282 |
+
|
| 283 |
+
def create_arg_parser():
|
| 284 |
+
""""Creates and returns the ArgumentParser object."""
|
| 285 |
+
|
| 286 |
+
# Instantiate the parser
|
| 287 |
+
parser = argparse.ArgumentParser(description=(
|
| 288 |
+
'embed.py creates ProstT5-Encoder embeddings for a given text '+
|
| 289 |
+
' file containing sequence(s) in FASTA-format.' +
|
| 290 |
+
'Example: python predict_3Di.py --input /path/to/some_AA_sequences.fasta --output /path/to/some_3Di_sequences.fasta --half 1' ) )
|
| 291 |
+
|
| 292 |
+
# Required positional argument
|
| 293 |
+
parser.add_argument( '-i', '--input', required=True, type=str,
|
| 294 |
+
help='A path to a fasta-formatted text file containing protein sequence(s).')
|
| 295 |
+
|
| 296 |
+
# Optional positional argument
|
| 297 |
+
parser.add_argument( '-o', '--output', required=True, type=str,
|
| 298 |
+
help='A path for saving the created embeddings as NumPy npz file.')
|
| 299 |
+
|
| 300 |
+
|
| 301 |
+
# Required positional argument
|
| 302 |
+
parser.add_argument('--model', required=False, type=str,
|
| 303 |
+
default="Rostlab/ProstT5",
|
| 304 |
+
help='Either a path to a directory holding the checkpoint for a pre-trained model or a huggingface repository link.' )
|
| 305 |
+
|
| 306 |
+
# Optional argument
|
| 307 |
+
parser.add_argument('--split_char', type=str,
|
| 308 |
+
default='!',
|
| 309 |
+
help='The character for splitting the FASTA header in order to retrieve ' +
|
| 310 |
+
"the protein identifier. Should be used in conjunction with --id." +
|
| 311 |
+
"Default: '!' ")
|
| 312 |
+
|
| 313 |
+
# Optional argument
|
| 314 |
+
parser.add_argument('--id', type=int,
|
| 315 |
+
default=0,
|
| 316 |
+
help='The index for the uniprot identifier field after splitting the ' +
|
| 317 |
+
"FASTA header after each symbole in ['|', '#', ':', ' ']." +
|
| 318 |
+
'Default: 0')
|
| 319 |
+
|
| 320 |
+
parser.add_argument('--half', type=int,
|
| 321 |
+
default=1,
|
| 322 |
+
help="Whether to use half_precision or not. Default: 1 (half-precision)")
|
| 323 |
+
|
| 324 |
+
return parser
|
| 325 |
+
|
| 326 |
+
def main():
|
| 327 |
+
parser = create_arg_parser()
|
| 328 |
+
args = parser.parse_args()
|
| 329 |
+
|
| 330 |
+
seq_path = Path( args.input ) # path to input FASTAS
|
| 331 |
+
out_path = Path( args.output) # path where predictions should be written to
|
| 332 |
+
model_dir = args.model # path/repo_link to checkpoint
|
| 333 |
+
|
| 334 |
+
if out_path.is_file():
|
| 335 |
+
print("Output file is already existing and will be overwritten ...")
|
| 336 |
+
|
| 337 |
+
split_char = args.split_char
|
| 338 |
+
id_field = args.id
|
| 339 |
+
|
| 340 |
+
half_precision = False if int(args.half) == 0 else True
|
| 341 |
+
assert not (half_precision and device=="cpu"), print("Running fp16 on CPU is not supported, yet")
|
| 342 |
+
|
| 343 |
+
seq_dict = read_fasta( seq_path, split_char, id_field )
|
| 344 |
+
predictions = get_3di_sequences(
|
| 345 |
+
seq_dict,
|
| 346 |
+
model_dir,
|
| 347 |
+
)
|
| 348 |
+
|
| 349 |
+
print("Writing results now to disk ...")
|
| 350 |
+
write_predictions(predictions,out_path)
|
| 351 |
+
|
| 352 |
+
|
| 353 |
+
if __name__ == '__main__':
|
| 354 |
+
main()
|
requirements.txt
CHANGED
|
@@ -2,3 +2,5 @@ dscript
|
|
| 2 |
biopython
|
| 3 |
pandas
|
| 4 |
tqdm
|
|
|
|
|
|
|
|
|
| 2 |
biopython
|
| 3 |
pandas
|
| 4 |
tqdm
|
| 5 |
+
transformers
|
| 6 |
+
sentencepiece
|